Unter COPD versteht man eine chronisch verlaufende Lungenerkrankung, die durch Entzündung im Bereich der kleinen Atemwege, Hypersekretion von Schleim, Obstruktion, Emphysembildung und Fibrosierung charakterisiert ist. Ausgelöst wird dies primär durch inhalative Noxen (z.B. Tabakrauch, Luftverschmutzung), bei Menschen < 50J auch durch einen Mangel an α1-Antitrypsin. Die Konsequenz ist in allen Fällen ein Überhang an Proteasen (Auflösung von Proteinen) mit Zerstörung der Lungenstruktur. Den akuten Asthmaanfall findet ihr hier. Beatmung findet ihr hier.
Symptome und Exazerbation
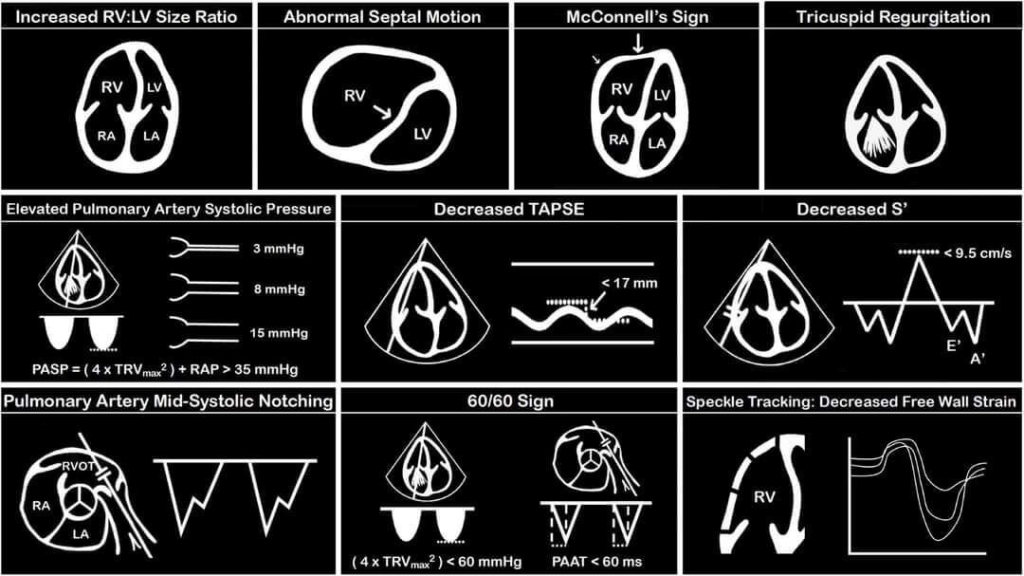
Der Symptomkomplex setzt sich primär aus Belastungsdyspnoe, Husten und schleimigem Auswurf zusammen. Auskultatorisch finden sich aufgrund der Bronchialobstruktion Giemen und Brummen. Therapeutisch wird daher insbesondere durch Ursachenbehebung (z.B. Rauchen ex), inhalative Bronchodilatatoren (β2-Mimetika wie Formoterol, Anticholinergika wie Ipratropium) und inhalative Steroide (z.B. Budesonid) eingegriffen. Das Auftreten einer akut exazerbierten COPD (AECOPD) ist gefürchtet und tritt va in den Herbst- und Wintermonaten auf. Diese ist durch Aggravierung der Symptomatik charakterisiert – es droht eine therapierefraktäre Bronchialobstruktion mit Atemversagen, da die Atemmuskulatur rasch erschöpft. Klinisch erkennbar ist dies an Sprechdyspnoe, angestrengter Atmung und ausgeprägter Zyanose unter maximalem Einsatz der Atemhilfsmuskulatur. Eine arterielle Blutgasanalyse zeigt initial zunächst Hypoxämie und Hypokapnie (Hyperventilation), mit zunehmendem Atemversagen letztlich eine Hyperkapnie bis hin zu dadurch bedingtem Koma. Es gilt anzumerken, dass COPDler aufgrund chronischer Limitierung des exspiratorischen Flusses mit unvollständiger Ausatmung und gestörtem Gasaustausch eher erhöhte arterielle CO2-Werte (bis 60 mmHg) „physiologischerweise“ zeigen – dies ist daher bei dieser Patientengruppe zunächst nicht allzu beunruhigend. Ultraschall ist von entscheidender Bedeutung, um nach Differentialdiagnosen zu suchen (z.B. Pneumonie, Pleuraerguss, Herzinsuffizienz), wobei die Differentialdiagnostik zur Pulmonalembolie (PE) erschwert ist (COPDler haben chronisch erhöhte rechtsventrikuläre Drücke, die im akuten Anfall drastisch erhöht sind und daher sonographisch zu Merkmalen einer akuten Rechtsherzbelastung führen können, z.B. McConnell-Zeichen, D-Zeichen, RV > LV, akute TI, TAPSE < 17 mm). Hilfreich ist hier der nachfolgende Graph, welcher Zeichen für eine PE zeigt. Immerhin ca 18% der AECOPD sind durch eine PE bedingt bzw. die eigentliche Ursache der Luftnot (Link).
Therapie
Therapeutisch hat sich initial folgendes Vorgehen bewährt:
- O2 (SpO2 bzw. SaO2-Zielwert 88-92%, hier)
- β2-Mimetikum p.i. (z.B. Salbutamol 5 mg)
- Anticholinergikum p.i. (z.B. Ipratropium 0,25-0,5 mg)
- Prednisolon p.o. oder i.v. (40 mg für mind. 5 Tage; siehe GOLD-Leitlinien)
Beachte, dass die inhalative Gabe von β2-Agonisten zu transienter Hypoxämie durch Aufhebung von HPV führen kann (β2 = Vasodilatation), Link, Link, Link. Kommt es zu keiner deutlichen Besserung innerhalb kurzer Zeit, so setze ich folgende Maßnahmen:
- β2-Mimetikum i.v. (z.B. Terbutalin in 0,1 mg Schritten – wirkt oft noch Wunder; die subcutane Gabe wird praktiziert, hat aber bei einer AECOPD nichts verloren, da man weder weiß wann und wie viel resorbiert wird -> S3 NVL Asthma spricht sich dagegen aus, S3 NVL AECOPD erwähnt Terbutalin s.c. nicht)
- Bronchodilatation über Magnesiumsulfat 2 g i.v. (Link)
- NIV-Therapie (IPAP 5-10 cm H2O, EPAP 3-5 cm H2O, Link, Link), dabei ev. Analgosedierung (Substanzen hier)
Absolute Kontraindikationen für NIV bei AECOPD: fehlende Spontanatmung oder Schnappatmung; massive, unbeherrschbare Sekretbildung; Atemwegsverlegung. Interessanterweise gibt es kaum Daten zu i.v. Anticholinergika bei AECOPD (obwohl diese ja p.i. gelistet sind!), wenn die inhalative Therapie refraktär ist, z.B. Glycopyrrolat oder Butylscopolamin. Ehrlicherweise würde ich bei schwerster AECOPD einen Therapieversuch probieren – das ist aber nur meine Empfehlung, die weder mit Daten noch Expertise belegt ist. Beachte, dass Theophyllin keinen Stellenwert mehr bei AECOPD spielt (Link, Link). Dasselbe gilt für Adrenalin – in der FOAM-Welt wird es zumindest angeschnitten und diskutiert. Bei schwerstem, therapierefraktärem, präletalem Bronchospasmus würde ich es zunächst i.m. und bei etwaigen Ansprechen i.v. geben (Link). Ein Antibiotikum ist bei massiver refraktärer Dyspnoe, zunehmendem oder purulentem Sputum oder Beatmungstherapie indiziert.
Kommt es auch unter dieser Maximaltherapie weiterhin zur Verschlechterung (zunehmende Bewusstseinstrübung bis hin zum Koma), so muss eine Notfallnarkose mit Intubation durchgeführt werden (Esketamin + Rocuronium unter Kreislaufunterstützung mittels Adrenalin oder Noradrenalin). Dabei ist auf eine suffizente Exspirationszeit durch lungenprotektive Beatmung (Tidalvolumen 6 ml/kg Idealgewicht, hoher Inspirationsflow, Atemfrequenz ~ 10, lange Exspiration, PEEP ~ 5 mmHg) zu achten, um die Bildung einer dynamischen Hyperinflation mit auto-PEEP, Barotrauma und Kreislaufzusammenbruch zu vermeiden. Insbesondere die Narkoseeinleitung von AECOPD oder Asthmaanfall kann zu Kreislaufstillstand aufgrund Sympathikolyse, Überdruckbeatmung, Rechtsherzversagen, Hypoxämie und Azidose führen und stellt daher eine maximal riskante Maßnahme dar. Genaue Details findet ihr im Asthmabeitrag (Link). Ein weiteres Problem ist das mögliche Auftreten eines Doppelpneus, der mit der invasiven Beatmung (bzw. bereits mit NIV-Therapie) zu spannen beginnt und dem Patienten endgültig den Rest gibt. Eine Fingerthorakostomie (mit ev. Bülau) ist dann beidseits nötig.
War da nicht noch was?
Ach ja genau, Atemstillstand durch zu hohe O2-Zufuhr. Ich wage zu behaupten, dass der Großteil der medizinisch tätigen Menschen dem Irrtum unterliegt, dass bei einem COPDler die Hypoxämie der wichtigste Atemantrieb ist und nicht mehr wie normalerweise die Hyperkapnie (s.o.). Deshalb würde eine zu hohe O2-Zufuhr mit Normalisierung des PaO2 die Chemorezeptoren „zufriedenstellen“, sodass keine atemantreibenden Reize mehr in die Medulla oblongata verbracht werden. Dies ist grundlegend falsch. Die wahren Gründe für Atemversagen bzw Apnoe durch Atempumpversagen durch zu hohe O2-Zufuhr bei COPD-Patienten sind (Link):
- Inhibition der hypoxisch-pulmonalen Vasokonstriktion (O2-Ansammlung in zerstörten Alveolarbezirken → Umverteilung von Blut in die nun gut belüfteten Alveolen → aufgrund Destruktion nur eingeschränkte Gasdiffusion → beeinträchtigte O2-Aufnahme und CO2-Abgabe → Hypoxämie und Hyperkapnie verstärkt)
- Reversierung des Haldane Effekts (→ desoxygeniertes Hämoglobin [Hb] bindet vermehrt CO2 → hohe O2-Zufuhr bedingt Oxygenierung des Hb mit Freisetzung von gebundenem CO2 bzw beeinträchtigter CO2-Bindung durch ebendieses oxygenierte Hb → Hyperkapnie)
Fallbericht
Vor kurzem erfolgte in meinem Dienst eine Parallelalarmierung von NEF und RTW zu einer bewusstlosen Person (75J, w). Die Patientin litt u.a. an COPD und war von Heimsauerstoff und 24h-Pflege abhängig. Sie schaffte Dinge des alltäglichen Lebens (Einkaufen, Körperpflege) mit Hilfe, wobei die Tochter selbst ebenfalls im Haus lebte. Seit der Früh war zunehmend ein redAZ aufgefallen, beim letzten Nachsehen war die Patientin dann bewusstlos. Weiters fiel der Pflege auf, dass die O2-Flasche leer sei. Die NFS des ersteintreffenden RTW erkannten ein kritisches A-Problem (Verlegung durch Schleim und Speichel -> zunächst gelöst durch Esmarch, Absaugen, Wendl – immer wieder nachrinnend). Erstmessung SpO2 60% (-> O2 15l/min). Bei Eintreffen NEF:
- A – dzt. mit obigen Maßnahmen offen, noch suffiziente Eigenatmung (jedoch zunehmend schwächer werdend)
- B – tiefe Hyperventilation, Tachypnoe ca 30/min, bdsts belüftet mit RG
- C -grenzwertig hypoton 105/65, HF 100, Rekap < 2s, Anlage IV Zugang x2
- D – GCS 5 dzt unklarer Genese, Pupillen PEARL mittelweit, BZ 100
- E – keine Verletzungszeichen erkennbar
Insgesamt lag also eine kritische Situation vor. Ich überstürzte sie aber nicht und informierte mein Team, dass ich zunächst mit der Tochter reden würde. Durch meinen intensivmedizinischen Background war für mich klar, dass ich hier nach Therapiebeschränkungen fragen musste. Tatsächlich legte mir die Tochter eine rechtsgültige und aktuelle Patientenverfügung vor (wieso sie das erst auf Nachfrage getan hatte war mir ein Rätsel), in welcher die Patientin u.a. lebenserhaltende sowie genau definierte intensivmedizinische Maßnahmen (Beatmung, CPR, Ernährung, Kreislauftherapie) bei irreversibler Erkrankung ablehnte. Ich erklärte der Tochter, dass ich präklinisch grundsätzlich nicht zwischen irreversiblen oder reversiblen Krankheitsbildern differenzieren konnte, da mir die diagnostischen Möglichkeiten fehlten (z.B. CT, Labor etc). Ich wies sie darauf hin, dass z.B. eine Exazerbation der Grunderkrankung, aber auch eine Sepsis, eine Hirnblutung oder eine Elektrolytstörung (z.B. HypoNa) ursächlich für den Zustand sein könnten. Deshalb klärte ich die Tochter über die Narkoseeinleitung auf, was sie verstand.
Ich kehrte zurück zu meinem Team und bemerkte, dass die Eigenatmung immer insuffizienter wurde (SpO2 97% – CAVE keine CO2-Messung!). Ich wies daher einen NFS an, eine assistierte Beatmung mittels Guedel durchzuführen, was toleriert wurde. Da wir uns im obersten Stockwerk des Hauses mit sehr enger Wendeltreppe befanden, war für mich die Sicherheit des Teams von oberster Priorität. Die Narkose würde ich sicher nicht im Zimmer durchführen, da mit dem Monitoring, der Beatmung und der Patientin eine ausgeprägte Sturzgefahr über die zweistöckige Wendeltreppe bestand. Deshalb schickte ich meinen NEF-Fahrer in den RTW voraus, wo er alles für die Narkoseeinleitung, Noradrenalin-Zufuhr und invasive arterielle Blutdruckmessung herrichtete, während die andern beiden NFS die Patientin einpackten. Gemeinsam trugen wir sie die Stiege hinunter. Beachte: die Patientin hatte bereits eine beginnende teilinsuffiziente Atmung. Ich nahm hier also das Risiko in Kauf, dass durch die ausbleibende assistierte Beatmung das CO2 weiter steigen würde, während wir sie hinuntertragen würden (Dauer ca 1 Minute). Sie hatte primär ja kein Oxygenierungs-, sondern ein Ventilationsproblem. Notfallmedizin ist nicht schwarz-weiß. Solche Aktionen darf man sich nur erlauben, wenn man Routine in der Behandlung kritisch kranker Patienten hat und Atemwegsmanagement einwandfrei beherrscht. Unten verbrachten wir sie auf die Trage und ich übernahm die assistierte Beatmung, während wir die Patientin in den RTW verluden.
Im RTW übernahm ein Kollege die assistierte Beatmung; eine Kapnographie zeigte Werte von 65 mmHg, cardiorespiratorisch war sie soweit stabil. Ich entschied mich daher noch für die ultraschallgezielte Anlage eines arteriellen Zugangs in die A. brachialis sin., was prompt und problemlos gelang. Auf einen Zugang schlossen wir laufendes Noradrenalin an (2 mg/50 ml). Nach Durchgehen der Intubationscheckliste erfolgte die Narkoseeinleitung mittels Esketamin 75 mg, Rocuronium 100 mg und Phenylephrin 0,2 mg i.v. und die Intubation erfolgte mittels VL Spatel 3, ID 7 und S-Guide. Die Kapnographie zeigte 75 mmHg, weshalb ich eine Hyperventilation durchführte. Achtung: Narkoseeinleitungen von COPD-Patienten sind gefährlich, da es durch den Wechsel in eine kontrollierte Beatmung zu Rechtsherzversagen mit Kreislaufstillstand kommen kann. Ein weiteres Risiko ist die Bildung von bilateralen Spannungspneus. Die weitere Narkose hielt ich mit Fentanyl und Midazolam aufrecht, der Kreislauf war mittels Noradrenalin stabil. Sie erhielt noch 2 g Magnesium i.v (Bronchodilatation). Sodann fuhren wir Richtung Spital. Vor dem Ausladen kam es zu einem SpO2-Abfall – in der etCO2-Kurve erkannte ich eine Haifischzacke, auskultatorisch war die Lunge spastisch – ich verabreichte daher noch Terbutalin 0,2 mg i.v., worunter es prompt zu einer Besserung kam. Die Übergabe erfolgte im SR an das Anästhesieteam. Eine CT-Diagnostik zeigte keine Hinweise auf ein intrazerebrales Geschehen, auch andere Ursachenfindungen waren ohne Ergebnis. Somit wurde am ehesten ein hyperkapnisches Koma in den Raum gestellt. Die Patientin wurde am Folgetag extubiert und entließ sich dann selbst von der Intensivstation. Sie verstarb kurze Zeit später.
Moin,
ich habe eine kurze Nachfrage zu der Sauerstoffinduzerten-Hyperkapnie bei COPD. Die HPV und der Haldane-Effekt habe ich verstanden. Aber warum resultiert aus der Hyperkapnie das Atempumpversagen?
Die Hyperkapnie führt zur sog. CO2-Narkose und einem ICP-Anstieg – eine zentrale Atemlähmung ist die Folge.