Unter Schock versteht man eine inadäquate Sauerstoffversorgung der Zellen. Dabei stellt die Zyanidintoxikation eine Sonderform dar, da die Zellen zwar ausreichend Sauerstoff angeliefert bekommen, diesen aber nicht nutzen können (Zyanid blockt die mitochondriale Atmungskette). Der Sauerstoff wird also einfach venös wieder abtransportiert – dies nennt man Arterialisierung des Blutes. Ganz allgemein kann man verschiedene Schockformen unterscheiden: distributiv, kardiogen, obstruktiv und hypovolämisch, wobei die Intoxikationen mit z.B. Zyanid, Propofol oder Metformin zum zytotoxischen Schock gezählt werden. Aufgrund des O2-Mangels kommt es zur Unterbrechung der aeroben Glykolyse – die Gewinnung von ATP kann nur mehr anaerob erfolgen; es entsteht Lactat als Endprodukt. Lactat ist für Zellen in diesem Zustand überlebensnotwendig, da seine Erzeugung Protonen puffert, die sich durch Blockade der Atmungskette (wo sie eigentlich eingeschleust werden sollen) anhäufen; mehr findet ihr unten im Beitrag. Die Schockazidose führt zu Pufferung der Protonen durch Bicarbonat; die Reaktion Proton (H+) + Bicarbonat (HCO3-) erzeugt Kohlensäure (H2CO3), welche in H2O und CO2 zerfällt. Der Patient im Schock hyperventiliert, um das anfallende CO2 abzuatmen bzw. mittels Senkung des arteriellen CO2-Gehaltes (PaCO2) die Reaktion von Proton und Bicarbonat zu beschleunigen.
Physiologie
Die O2-Versorgung des Körpers beruht auf der Transportkapazität des Hämoglobins (Hb) sowie zu einem sehr kleinen Teil auf in Blut gelöstem O2. Es gilt: O2-Konzentration (CO2) im Blut = HbO2 + gelöster O2. HbO2 = Hb x SaO2 x 1,34 (sog. Hüfnerzahl, d.h. 1 g Hb kann 1,34 ml O2 binden), während sich die Menge des gelösten O2 aus PO2 x 0,03 (Löslichkeitskoeffizient) ergibt. Es gilt: CO2 (ml O2 /dl Blut) = (Hb x SO2 x 1,34) + (PO2 x 0,003). Für arterielles Blut (CaO2) gilt ein Normwert von ca 20 ml O2 /dl Blut (zur Berechnung einfach die Werte aus einer aBGA eingeben), venös (CvO2) 15 ml O2 / dl Blut. Es ist erkenntlich, dass nur 25% des O2 von Zellen tatsächlich genutzt werden. Die übrigen 75% stellen also eine Reserve dar, falls plötzlich akut mehr O2 benötigt wird. Die sog. zentralvenöse Sättigung (ScvO2), welche man aus einem ZVK bestimmen kann, beträgt ungefähr 70% und kann daher als Surrogatmarker für eine zelluläre Unterversorgung genutzt werden (beträgt ScvO2 z.B. 50%, so extrahieren die Zellen deutlich mehr Sauerstoff – es gibt also ein Problem auf zellulärer Ebene). DO2 ist das O2-Angebot im Gesamtblut, VO2 der O2-Bedarf. DO2 errechnet sich aus HZV (l/min) x CaO2 (ml/dl) x 10 (Umrechnungsfaktor, da HZV in l, CaO2 in dl angegeben ist), d.h. 5 x 20 ml x 10= 1000 ml O2/min. Der Netto-VO2 beträgt also 250 ml O2/min (25%).
Studieren wir die CO2-Formel weiter so ist ersichtlich, dass wir bei Hypoxie primär nur den Hb und die O2-Sättigung (durch O2-Gabe) manipulieren können, um die O2-Transportkapazität zu erhöhen. Das heißt nicht, dass wir in hypoxische Patienten massenhaft Erykonserven reinschütten (denn diese haben signifikante Komplikationen bei undurchdachter Gabe), sondern dass wir z.B. bei einem Patienten im hämorrhagischen Schock mit Hb 3 g/dl jedenfalls Erys zuführen müssen, da durch den erheblichen Blutverlust ein relevanter Anteil an Erys aus dem Gefäßsystem ausgetreten ist. Den Anteil des gelösten O2 kann man in einer hyperbaren Druckkammer (siehe Tauch- oder CO-Unfall) relevant erhöhen, da eine künstliche Erhöhung des atmosphärischen Drucks die Löslichkeit des Sauerstoffs verbessert (Gesetz nach Henry) und wir dadurch Gewebe suffizient versorgen können. Aber: ein COPD-Patient mit Hb 10 g/dl und schwerer Hypoxämie ist keine Indikation für eine Blutkonservengabe, auch wenn es irgendwie logisch erscheint! Das Nebenwirkungsprofil und die begrenzte Verfügbarkeit der Konserve überwiegen den therapeutischen Effekt.
Lactat
Die Lactatazidose ist etwas, das es gar nicht gibt. Biochemisch möchte ich mich hier auf das absolute Minimum beschränken. Eine großartige Aufarbeitung findet ihr hier und hier. Leider werden seit Jahrzehnten von Experten und auch in Lehrbüchern zwei grundlegende biochemische Aspekte verwechselt: Milchsäure und Lactat. Sieht man sich folgende Abbildung an, so wird ersichtlich, dass sie nicht dasselbe sind. Lactat ist das Anion (Base) der Milchsäure, das heißt Milchsäure gibt ihr Proton ab (daher Säure) und es entsteht Lactat. Lactat ist also gar nicht in der Lage, ein Proton abzugeben und so eine Azidose zu erzeugen. Aber was ist Lactat dann? Grundsätzlich kann eine Zelle auf zwei Wegen lebensnotwendiges Adenosintriphosphat (Energiequelle) generieren: aerob (O2 muss verfügbar sein) und anaerob (bei zellulärer Hypoxie; Endprodukt: ATP und Lactat). Somit ist Lactat ein Stoffwechselendprodukt der anaeroben Glykolyse (das heißt aus Glukose wird ATP generiert). Diese läuft sehr schnell ab und generiert rasch ATP unter Stress bzw. hypoxischen Zuständen. Diese ATP-Generierung ist überlebenswichtig für die Zelle. Lactat kann ultimativ zur Glukoneogenese verwendet werden, was u.a. im Herzen geschieht. Interessanterweise gibt es mittlerweile Arbeiten, die exakt aus diesem Grunde eine therapeutische Anwendung von Lactat in den Raum stellen (Link), um z.B. das Outcome von kritisch kranken Patienten zu verbessern (Energiesubstrat, Antiinflammation). Von einem schädlichen Stoff kann also keineswegs die Rede sein.
Es werden unterschiedliche Formen der Hyperlactatämie unterschieden. Typ A beruht auf einem tatsächlichen zellulären O2-Mangel bzw. der Unmöglichkeit Sauerstoff in der Atmungskette (sog. oxidative Phosphorylierung in den Mitochondrien) zu verwerten (z.B. Intoxikation mit Blausäure, welche zur Blockade der Atmungskette führt). Auch im Rahmen einer Sepsis können Toxine die Atmungskette blockieren und so die Lactatspiegel erhöhen, da der Körper „gezwungen“ ist, die anaerobe Glykolyse zur ATP-Generierung durchzuführen. Selbiges gilt für das PRIS durch Propofol. Typ B beruht auf erhöhter Generierung von Lactat (z.B. Ankurblung des zellulären Stoffwechsels durch Beta-Mimetika, siehe prolongierte Inhalation von Salbutamol bei aeCOPD bzw. Asthma oder Adrenalin bei Anaphylaxie) oder reduzierter Lactat-Clearance (z.B. Leberversagen). Diese Arbeit teilt die Hyperlactatämie überhaupt neu ein:
- Überschuss an Pyruvat (Alkalose, adrenerge Agonisten, Tumore, Alkohol, diabetische Ketoazidose)
- reduzierte Verstoffwechselung von Pyruvat (Hypoxie, Schock, CO, Cyanid, PRIS, Metformin)
- reduzierte Lactat-Clearance (Leber- oder Nierenversagen)
Was bedeutet das alles jetzt? Zunächst muss man zwei Punkte verstehen: Milchsäure ist nicht Lactat und Milchsäure existiert auch in keinem Lebewesen (Link), denn sie dissoziiert (wenn wir sie z.B. mit dem Essen zuführen) stets. Vom Körper selbst wird Milchsäure auch nicht produziert (Link). Biochemisch ist zunächst zu verstehen, dass NAD+ ein intrazellulärer Elektronentransporter ist und diese Elektronen als NADH (durch Annahme von einem Proton und zwei Elektronen wird NAD+ zu NADH reduziert) an die Atmungskette (Link, Link) übergibt. Dadurch wird der Ablauf der Atmungskette überhaupt erst ermöglicht. Und Lactat? Im Rahmen der Glykolyse entstehen 4 Protonen und zur Kompensation von 2 Protonen wird 2x NAD+ zu 2x NADH reduziert (s.o.), die übergebliebenen 2 Protonen werden entweder in die funktionierende Atmungskette eingeschleust oder durch Umwandlung von 2 Pyruvat zu 2 Lactat verbraucht. Es gilt: 2 Pyruvat + 2 NADH + 2 H(+) → 2 Lactat + 2 NAD(+).
Moment mal. Wo kommen dann freie Protonen her die eine Azidose verursachen können? Ganz einfach. Wird ATP zu ADP hydrolysiert, entstehen freie Protonen, welche in der Folge dann in die Atmungskette eingeschleust werden. Es gilt: ATP + H2O → ADP + H(+) + Pi (anorganisches Phosphat). Ist die Einschleusung der Protonen in die Atmungskette nicht möglich (z.B. weil sie blockiert ist), kommt es zur Akkumulation und Azidose (Link). Parallel läuft natürlich die anaerobe Glykolyse mit Generierung von Lactat ab (was Protonen verbraucht, s.o.!). Das bedeutet der Lactatanstieg ist Begleitphänomen einer Azidose, nicht die URSACHE, und dient in gewissem Ausmaß der Pufferung der Azidose! Veranschaulicht wird dies in diesem Bild (Quelle):
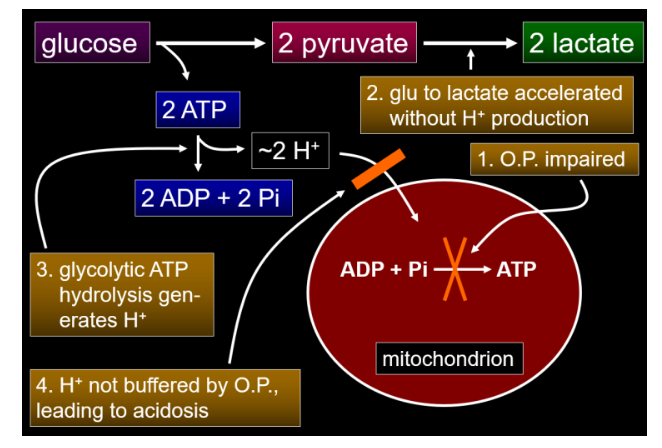
Mit einer Hyperlactatämie (> 2 mmol/l) zeigen uns die Zellen meist also an, das irgendetwas nicht stimmt, beispielsweise die zelluläre O2-Versorgung oder -Nutzung. Die Anhäufung von Lactat garantiert das vorübergehende Überleben der Zelle, da es ATP generiert, als Energiesubstrat dient (ua Glukoneogenese) und nach Behebung der Ursache wieder zu Pyruvat umgewandelt werden kann (und dieses kann wiederum in die erneut funktionierende Atmungskette zur ATP-Generierung eingeschleust werden). Eine Lactatazidose existiert nicht und eine Umbenennung in Azidose mit Hyperlactatämie scheint ratsam, um weitere Verwirrungen zu unterbinden.
Praktisches Vorgehen bei Schock
Zunächst ist wichtig zu verstehen, dass Schock nicht Hypotonie ist. Bei Hypoperfusion von Zellen kommt es zunächst zu einer kompensatorischen Stressreaktion des Körpers, d.h. ausgeschüttete Hormone (Katecholamine, RAAS, Vasopressin, Cortisol) führen zu einer Blutdruckstabilisierung und Tachypnoe (erhöhte O2-Aufnahme, erhöhte O2-Abgabe). Es ist also trügerisch zu denken, dass ein Patient mit normalen Vitalwerten keinen Schock aufweist. Entscheidend ist hier die Suche nach Markern der Hypoperfusion bzw. des zellulären Stress, nämlich Agitation, unerklärliche Tachykardie, kalt-blass-zyanotische Haut, Rekap-Zeit > 2s, Oligurie, aber auch apparative Werte wie ScvO2 < 70%, Bicarbonatmangel oder Lactatanstieg. Eventuell ist eine metabolische Azidose erkenntlich. Das weitere (therapeutische) Vorgehen richtet sich ganz grundsätzlich nach den verschiedenen Ursachen des Schocks (wobei Kombinationen vorliegen können, z.B. kann ein schwer verletzter Patient hypovolämische und neurogene Schockformen aufweisen). Die Behebung der Ursache ist die Priorität (z.B. PCI bei OMI, Link). Überbrückend können Vasopressoren und Inotropika eingesetzt werden, siehe hier, hier und hier.
Beispiele:
- hämorrhagischer Schock: Blutungskontrolle, permissive Hypotonie (Normotonie bei SHT oder RMT), Massentransfusion (häufig als 4:4:1 Schema, d.h. 4x Erys + 4x Plasma + 1x Thrombos), Wärmeerhalt, Gerinnungsfaktoren (empirisch häufig 1-2 g TXA, 2-4 g Fibrinogen, 3 Ampullen Calciumgluconat oder 1 Ampulle Calciumchlorid, 2000 Einheiten Prothrombinkonzentrat PPSB; idealerweise erfolgt die Gabe ROTEM gesteuert)
- 2 g TXA bei massiver Hämorrhagie wird explizit von NAEMSP, TCCC und Levy et al. empfohlen (s. oben)
- PPSB enthält Prothrombin FII + Prokonvertin FVII + Stuart-Faktor FX + antihämophiler Faktor B FIX + Proteine C / S / Z + Heparin zur Minimierung des Thromboserisikos (gegen Clotting in der Verpackung) – eine Alternative ist FEIBA (wie PPSB, nur ohne Heparin -> nutzen bei Anamnese von Heparin-induzierter Thrombozytopenie II)
- anaphylaktischer Schock: siehe hier
- septischer Schock: siehe hier
- kardiogener Schock: siehe BRASH (Link); weiters Ursache
- Lungenembolie: Heparin (NMH als 1. Wahl gelistet in rezenter PE-Leitlinie, Link), Lyse, Thrombektomie, symptomatisch – CAVE Narkoseeinleitung (Link) und Intubation (Link) kann zu Rechtsherzversagen führen
- Pericardtamponade: ultraschallgezielte Nadel- oder chirurgisch-operative Entlastung (CAVE Narkoseeinleitung und Intubation kann zu Kreislaufzusammenbruch führen; Adrenalinboli fraktioniert; Einleitung nur in Schnittbereitschaft des Herzchirurgen)
- neurogener Schock: Vasopressoren, ev. Flüssigkeit – CAVE nicht mit spinalem Schock verwechseln (vollständiger Verlust der Rückenmarksfunktion, also z.B. keine Motorik, keine Sensibilität etc), Operation
- Zyanid: Hydroxocobalamin (1. Wahl), alternativ 4-DMAP, Natriumthiosulfat, Dicobaltedetat
- CO: O2
- usw.
Sonderfall hämorrhagischer Schock
Im hämorrhagischen Schock setze ich v.a. bei desaströsen Venen sehr gerne einen orangenen Zugang (14G) ultraschallgezielt in die linke V. jugularis interna (dies ist in der FOAM-Welt als „Easy Internal Jugular Vein“ bekannt), um unverzüglich einen großlumigen Zugang zum Schütten zur Verfügung zu haben und erste Blutkomponenten verabreichen zu können. Hagen-Poiseuille waren zwei sehr intelligente Menschen, die festhielten, dass die Durchflussmenge einer Flüssigkeit durch ein Rohr unter anderem von Flüssigkeitsviskosität, Länge und Durchmesser des Rohrs abhängt. Heißt im Klartext: kurze, fette Zugänge (14G) sind ideal für Transfusionen. Ein herkömmlicher ZVK hat nur eine Flussrate von maximal einem grünen Venenzugang, da der ZVK einfach zu lange dafür ist. Die rasche Einlage eines 14G Zugangs, der Flussraten bis 350 ml/min erreicht, ermöglicht eine schnellere Stabilisierung des Patienten als das Auftischen und sterile Einbringen eines DIAK. Wenn Zeit besteht kümmere ich mich um weitere ultraschallgezielte 14G Zugänge (Cubita, Basilicavenen) bzw. setze dann gleich einen Dialysekatheter (DIAK) in die rechte Jugularvene.
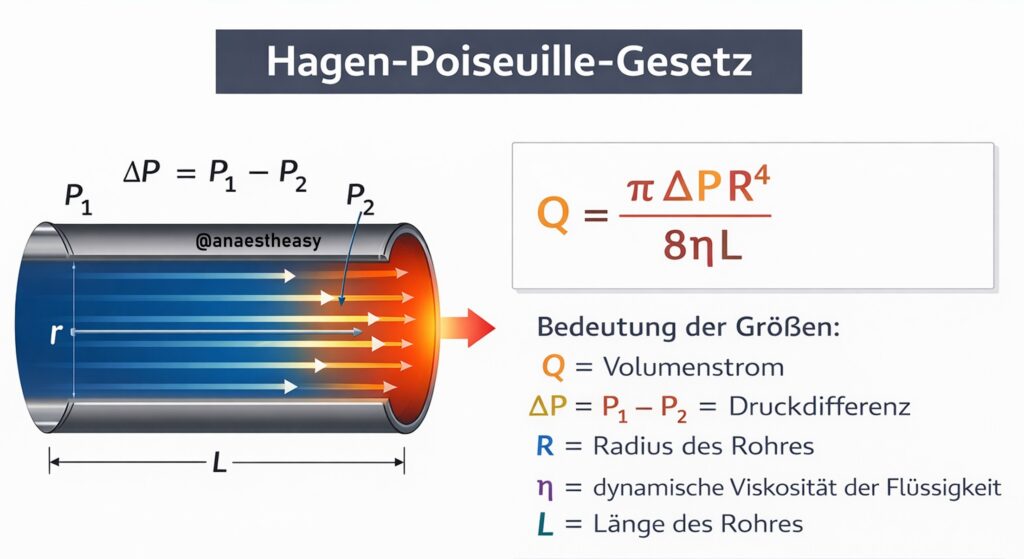
Merke: die aggressive Flüssigkeitsgabe (Kristalloide) hat keinen Stellenwert bei akut und unkontrolliert blutenden Patienten bzw. Massentransfusion, da es zu Hypothermie, Verdünnung von Gerinnungsfaktoren und Aufreißen des Clots kommt und Kristalloide keine O2-Transportkapazität besitzen. Ich verabreiche daher so gut wie kein Kristalloid, was u.a. mit TCCC 2026 (Link) bzw der Leitlinie von Levy et al (Link) übereinstimmt. Die Europäische Traumaleitlinie empfiehlt max. 1000 ml (Link). Erst wenn die Blutung operativ gestillt worden ist verabreiche ich langsam gewärmte Kristalloide, behalte dabei aber den Hämoglobinwert und die Hämodynamik engmaschig im Auge. Verliert ein Patient initial nur Blut und wird dieses nicht ersetzt, so ist der Hb zunächst immer ident, d.h. selbst wenn jemand 3 Liter Blut verloren hat, so wird eine BGA einen unveränderten Hb-Wert zeigen. Erst mit Zufuhr von Flüssigkeit kommt es zum Verdünnungseffekt, welcher dann den Hb-Wert verdünnt, weil 3 Liter Blut nicht durch 3 Liter Blut, sondern 3 Liter Kristalloid ersetzt worden ist (d.h. das restliche Hb verdünnt sich dann nicht mehr reine 6 Liter Blut, sondern 3 Liter Blut und 3 Liter Kristalloid – der Wert stürzt ab). Wie dumm das ist kann man sich selbst ausmalen. Merke: Patienten bluten Blut, keine Kristalloide.
Eine Ausnahme sind Patienten, die Blut in solch einer Menge verloren haben, dass keine akute Lebensgefahr resultiert, und bei denen die Blutung rasch gestillt werden konnte. Auch Patienten im kompensierten Schock mit rascher Blutstillung können zunächst einer vorsichtigen Kristalloidtherapie zugeführt werden. Ein Beispiel aus meinem Alltag ist ein Patient, welcher sich in suizidaler Absicht beidseits die Radialarterien aufgeschnitten hat und von der Notärztin im präletalen Zustand vorgefunden wurde. Der Blutverlust wurde auf 1-2 Liter geschätzt. Die Blutungen wurden beidseits mittels Israeli-Bandage gestillt. Sie verabreichte einen halben Liter Kristalloid und 0,1 mg Phenylephrin, was zu einer transienten Besserung des Patienten führte. Im Schockraum präsentierte sich mir ein Patient mit GCS 15, HF 140/min und RRsys 90 mmHg. Unverzüglich setzte ich ultraschallgezielt einen arteriellen Zugang in die A. radialis rechts (Mitte des Unterarms; brachial beidseits keine Möglichkeit weil Venenzugänge in Cubita liegend und distal radial nicht möglich wegen der Verletzungen), was mir unter mm-Arbeit auch gelang (massive Vasokonstriktion bei Schock). Immerhin habe ich in meiner Karriere bereits > 400 Arterien ultraschallgezielt gesetzt, sodass es bei mir funktioniert, wenn es darauf ankommt. Die erste BGA zeigte einen pH von 7,2, Lactat 15, BE -18 und Hb 10 g/dl. Der Patient hatte also ein signifikantes Schockgeschehen. Da jedoch die Blutung zu dem Zeitpunkt kontrolliert war verabreichte ich zunächst 2 Liter EloMel und 1 Liter Gelofusin, wohlwissend dass eine Erythrozytentransfusion aufgrund Hämodilution in der nächsten Stunde notwendig sein würde. Hier ist es vertretbar, den Blutverlust vorsichtig mit Kristalloiden zu behandeln und durch den Verdünnungseffekt eine Hämodilution zu initiieren, d.h. das verlorene Blut wird durch Kristalloide ersetzt, was zu einem Abfall des Hämoglobinwertes durch Verdünnung führt (z.B. von 13 g/dl auf 8 g/dl). Da der Patient unter aHT und DM litt setzte ich meine Transfusionsgrenze bei 7 g/dl an. Zur operativen Versorgung durch Unfallchirugie / Gefäßchirurgie entschied ich mich dann für die beidseitige Anlage eines AxPlex mit insgesamt 40 ml Ropivacain 0,5% und 0,3 mg Clonidin, dazu Sedierung mit Dexmedetomidin und Esketaminboli. Eine Narkose ist hier auf jeden Fall zu vermeiden, da es zur Dekompensation bis hin zu Kreislaufstillstand kommen kann. Eine Regionalanästhesie ist hier einfach die eleganteste Wahl und zeigt wie facettenreich unser Fach ist. Weiters sorgt die Plexublockade für eine Vasodilatation im Arm, was die Mikroperfusion erheblich verbessert. Er erhielt von mir noch einen orangenen Zugang ultraschallgezielt in die V. basilica dx. Die Kontrollgase während der OP zeigten eine zunehmende Besserung der Zellperfusion, der Hb sank aber parallel auf 6,9 g/dl und ich entschied mich zur Gabe von 2x Erythrozytenkonzentraten. Weiters verabreichte ich ROTEM-gesteuert 1 g Tranexamsäure und 4 g Fibrinogen. Der Patient überstand die OP gut, eine Gefäßrekonstruktion war überraschenderweise nicht notwendig.
Wichtig bei Patienten im hämorrhagischen Schock ist auch die Frage nach Blutverdünnern, sodass sich auch hier das Management mit Antidota ändert:
- ASS, ADP-Blocker: Desmopressin (V2-Agonist, fördert Freisetzung von vWF und FVIII aus Endothel) -> beachte, dass Ticagrelor Thrombozyten nur reversibel bindet und dadurch bei Thrombozytentransfusion die neuen Blutplättchen ebenfalls inhibiert
- FXa-Hemmer (Apixaban, Rivaroxaban, Edoxaban): PPSB, alternativ Plasma oder Andexanet Alfa (keine Zulassung mehr in USA, da kein Vorteil ggü. PPSB bzgl. Kosten und Effektivität)
- FIIa-Hemmer (Dabigatran): Idarucizumab, alternativ PPSB
- Vit-K-Blocker (Cumarine wie Phenprocoumon): PPSB + Vitamin K, alternativ FEIBA oder Plasma
- unfraktioniertes Heparin (UFH), niedermolekulares Heparin (NMH, s. Enoxaparin): Protamin (UFH 100%, NMH 50-80%)
- Fondaparinux, Danaparoid (langwirksame synthetische Heparine): kein Antidot verfügbar; PPSB oder Plasma
- Bivalirudin und Argatroban sind kurz wirksame Thrombinhemmer (FII); meist reicht eine Beendigung der Infusion zur Reversierung der Gerinnungshemmung
- weitere therapeutische Optionen sind FXIII und FVII (Ultima Ratio)
Exemplarisch sei der PPH-Algorithmus aufgelistet (Link), welcher für uns Anästhesisten im Kreißsaal eine ganz besondere Bedeutung hat, siehe Sectio.
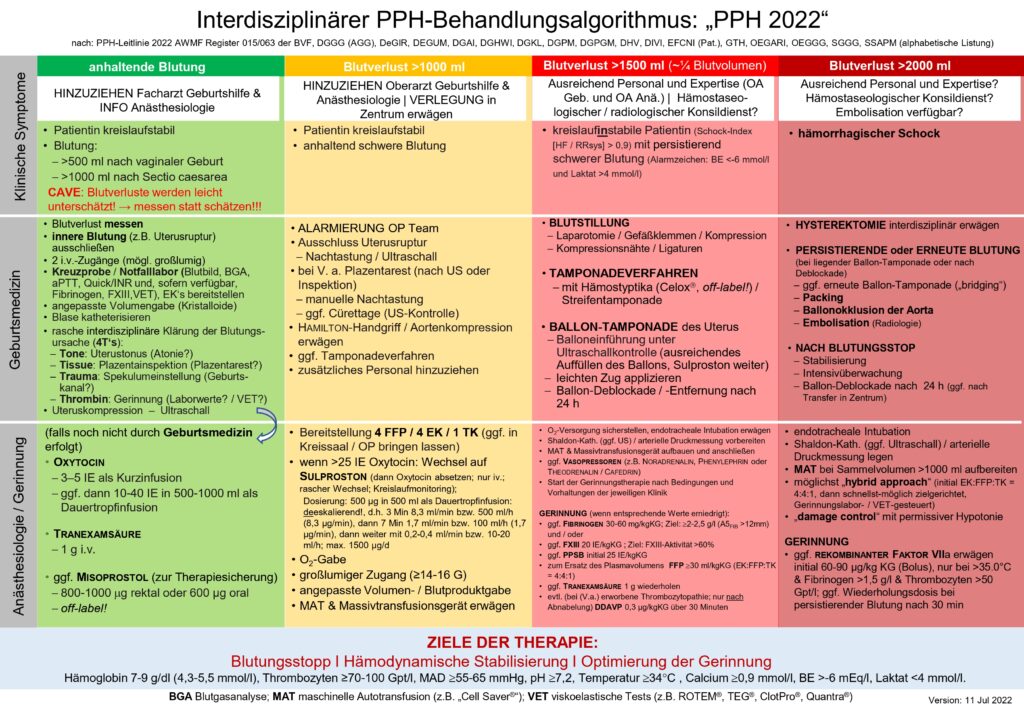
Beachtet, dass hier u.a. die REBOA erwähnt ist (mehr im Beitrag Reanimation der Zukunft hier). Innerklinisch sind mir mit REBOA zwei Massentransfusionen im Kopf geblieben (einmal nach massiver Blutung aus Lebergefäßen und oberem GIT mit SALAD-Intubation, einmal nach rupturiertem infrarenalem Aortenaneurysma). Das rupturierte AA war die heftigste Massentransfusion war die ich in meiner Laufbahn durchgeführt habe. Der Patient kam präletal von der Intensivstation in den OP, intermittierend Herzdruckmassage, 20 mg (!!) Noradrenalin auf 50 ml verdünnt mit Laufrate 100 ml/h über einen femoralen ZVK, zusätzlich Vasopressin. Während eine Oberärztin die Koordination durchführte platzierte ich einen Dialysekatheter in die V. jugularis interna, parallel schwemmte der Gefäßchirurg einen REBOA-Katheter ein und ich platzierte zusätzlich noch links einen weiteren ZVK in die V. jugularis interna. Der Chirurg und ich brauchten insgesamt 5 Minuten für diese Aktion. Während er den Bauch laparotomierte schütteten wir über ein Rapid Infusion System hinein was wir konnten. Es ist echt irre was eine Flussrate von 0,5 l/min bei einem Dialysekatheter ermöglicht (Hagen-Poiseuille lassen grüßen): Adrenalin inkl. Perfusor, Noradrenalin, Vasopressin, 12x Erythrozytenkonzentrat, 8x Fresh Frozen Plasma, 3x Thrombozytenkonzentrate, 16 g Fibrinogen, 4000 IE PPSB, 2 g Tranexamsäure, 3A Calciumglukonat. Trotzdem blieb der Hb stetig < 3,5 g/dl. Das ROTEM war ein Totalausfall, chirurgisch war eine inoperable Dissektionslamelle erkenntlich, welche auch Seitenäste erfasste. Erschwerend kam noch hinzu, dass von einer vorherigen CT-Diagnostik ein Pneumothorax links beschrieben worden war. Da mein Beatmungsdruck (Link) intra-OP sukzessive von PIP 22 auf PIP 31 anstieg und der Patient mit der O2-Sättigung (CAVE schwere Anämie) nicht anstieg führte ich im Zweifel noch eine Fingerthorakostomie links mit Bülaudrainageneinlage durch. Unsere Rettungsversuche waren letztlich aber aussichtslos und wir brachen die OP ab. Der Fall zeigt wieder mal eindrucksvoll, wie spannend, herausfordernd, belastend und maximalinvasiv das Fach Anästhesiologie eigentlich ist.
ROTEM
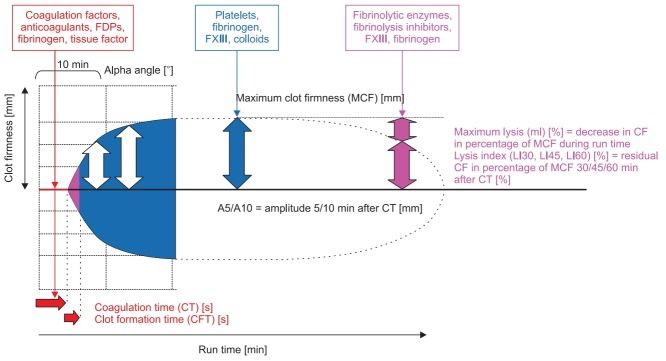
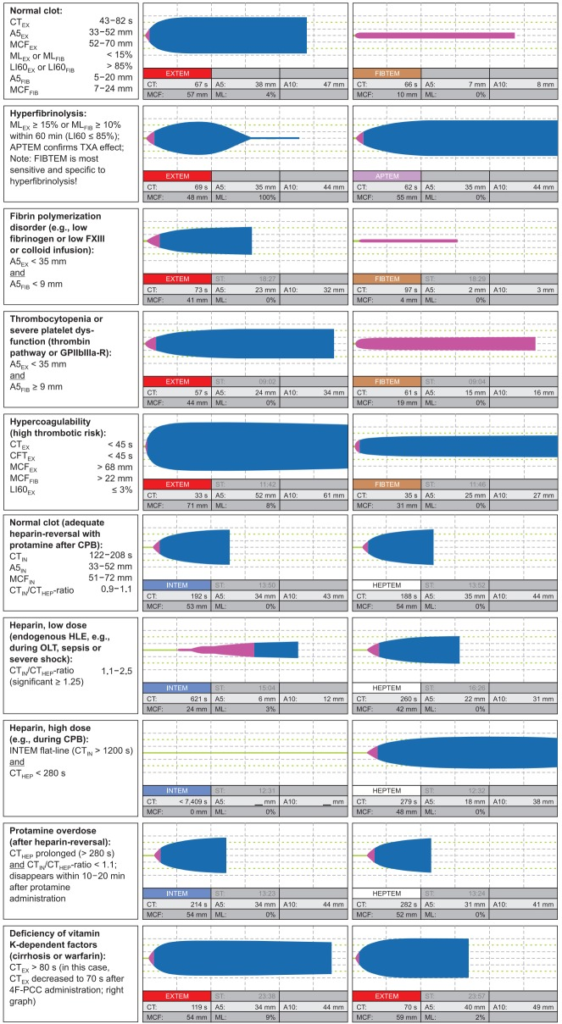
Wie bereits oben angesprochen sind anerkannte Schemata bei Massentransfusion entweder 1:1:1 (Ery : Plasma : Thrombos – Link) oder 4:4:1 (Ery : Plasma : Thrombos – Link). Flankiert wird das häufig noch durch 2-4 g Fibrinogen, 1-2 g Tranexamsäure, Calcium (3A CaGlu, 1A CaCl) und – falls das Auftauen des Plasma zu lange dauert – 2000 Einheiten PPSB. Das ROTEM (Rotationsthrombelastometrie) ist von essenzieller Beurteilung, wenn es um die gezielte Gerinnungssubstitution geht. Zwar verabreichen wir bei Patienten im hämorrhagischen Schock häufig empirisch bereits ein Arsenal an Blutprodukten bzw. Gerinnungsfaktoren, jedoch drohen hier bei unreflektierter Gabe lebensbedrohliche Nebenwirkungen wie Blutgruppeninkompatibilität, Anaphylaxie, Infektionen, Lungenschädigung (TRALI) oder Lungenödem (TACO). Nach unserem Erstangriff (s.o.) sollten wir ein ROTEM abnehmen (wenn noch keines zuvor erfolgt ist) – die Auswertung benötigt 10 Minuten. Wir können vergleichen, was unsere Therapie gebracht hat und wo wir noch optimieren können. Das ROTEM ist für uns Anästhesiologen von entscheidender Bedeutung, sei es im OP, auf der Intensivstation oder im Schockraum. Im Kern wird beobachtet, wie der Clot zustande kommt. Kernelemente sind:
- Clotting time (CT; Zeit von Start bis 2 mm oberhalb Basislinie) → Initiierung Gerinnungskaskade
- Clot formation time (CFT; Zeit von 2 mm bis 20 mm) und α angle → Kaskade verstärkt
- A10/A30 = Amplitude nach 10/30 Min
- Maximum clot firmness (MCF) = Thrombozytenaggregation und Thrombusstärke
- Lysis index (LI) z.B. nach 30 Min → Lyse des Thrombus
- exTEM (extrinsischer Gerinnungsweg, Tissue Factor als Trigger)
- inTEM (intrinsischer Gerinnungsweg, Phospholipide und Ellagsäure als Trigger)
- fibTEM (extrinsischer Gerinnungsweg, Einfluss von Thrombos durch Cytochalasin D gehemmt → Funktionalität und Menge von Fibrinogen überprüft)
- apTEM (extrinsischer Gerinnungsweg mit Hinzufügen von Aprotinin zur Beurteilung der Fibrinolyse)
- hepTEM (intrinsischer Gerinnungsweg mit Zusatz von Heparinase zur Hemmung von Heparineffekten)
Die initiale Clotbildung kann v.a. durch Antikoagulanzien oder Mangel an Gerinnungsfaktoren beeinträchtigt sein, während Defizite in einem bereits entstandenen Clot primär auf Thrombozyten- oder Fibrinogenmangel, Hyperfibrinolyse oder Mangel an FXIII (wichtig für Festigkeit des Clots) bedingt sind. Eine großartige Übersicht findet sich hier: